Талассемия – это патология крови, для которой характерны нарушения в формировании одной или сразу нескольких цепей гемоглобина в клетках эритроцитов. Это становится причиной развития малых, некачественных эритроцитов.
При таком заболевании наблюдается ухудшение транспортировки кислорода к тканям и клеточным элементам организма. Это вызывает их гипоксию, становятся очевидными проявления признаков болезни.
Раньше недуг чаще наблюдался в климатически жарких местах, где люди страдают малярией: Юго-Восточной Азии, Африке, странах Средиземноморья. Но в современном мире он диагностируется в самых разных регионах из-за миграции населения. Патология формируется одинаково у лиц женского и мужского пола.
.jpg)
Ежегодно рождается около 200 000 детей с этой болезнью, у 50% из них диагностируется гомозиготная форма β-талассемии. Последняя быстро приводит к смерти.
При гетерозиготной форме болезнь себя не проявляет, это возможно только в экстремальных ситуациях. Однако высок риск ее развития у потомков, они могут получить ее как наследственную патологию.
Причины талассемии
Они изучены хорошо, поскольку заболевание достаточно распространено. Достоверно известно, что первые изменения наблюдаются при нарушении синтеза белковых цепей гемоглобина, который способен связываться с молекулами кислорода и транспортировать их ко всем тканям организма.
Чаще всего свое развитие заболевание получает из-за генов, которые дети наследуют от своих родителей. В тех из них, которые несут ответственность за синтез гемоглобина, обнаруживается мутация.
При изучении структуры обнаруживается пигмент, в состав которого входит железо и 2 пары соединений белка: альфа- и бета-цепи. Если синтез одной из них нарушается, то накапливается вторая.

Провоцируют такие изменения те гены, что несут ответственность за формирование белка гемоглобина. При такой мутации идет нарушение правильного набора аминокислот в цепочке. Явление характерно и для возбудителя малярии.
Наследственность у ребенка способна проявиться в двух вариантах:
- Гомозиготный вид патологии – от мамы и папы одновременно.
- Гетерозиготный – от одного из родителей.
Классификация
Если аномальный белок гемоглобина наследуется с обеих родительских сторон, то это тяжелая форма, для которой характерны ярко выраженные проявления. Это большая талассемия, или болезнь Кули (в честь доктора, что ее описал).
.jpg)
Она наследуется по типу рецессивному (двухаллельная система), в основе лежит снижение синтеза цепей полипептидов.Если это идет со стороны одного родителя, то при отсутствии крайне неблагоприятных обстоятельств признаки отсутствуют.
В современной классификации выделены следующие формы недуга:
- Альфа-талассемия. Наблюдается при отсутствии одного либо нескольких генов, которых в норме должно быть по два от матери и отца. Именно такое количество обеспечивает достаточную выработку белков цепочки альфа-глобина. При отсутствии двух генов возможно проявление симптомов анемии. При недостатке трех генов диагностируется гемоглобинопатия H, которая ярко выраженно проявляется симптомами малокровия. Отсутствие четырех генов – явление довольно редкое, которое носит название “водянка плода”. Такие дети умирают до, во время либо сразу после родов.
- Бета-талассемия наблюдается при нарушении одного либо двух генов, которых в норме идет по одному от папы и мамы. Вырабатывается недостаточное количество белка бета-глобина. При видоизменении одного гена человек является носителем болезни, не исключено проявление умеренного малокровия. При изменении обоих генов одновременно диагностируется бета-талассемия интермедия (анемия Кули). При основной развивается и тяжелая форма анемического синдрома. Гомозиготная бета-форма выявляет себя к году жизни ребенка и различается проявлением признаков. Гетерозиготная (малая форма талассемии) характеризуется размытыми симптомами, либо признаки полностью отсутствуют.
Нужно учитывать, что в отдельных случаях гомозиготная форма протекает легко, ее нельзя квалифицировать как большую талассемию, поэтому ее отмечают как промежуточную.
У этой патологии есть три степени:
- Тяжелая. Аномальные изменения настолько сильны, что ребенок умирает на первом году жизни.
- Среднетяжелая. Ребенок способен прожить около восьми лет.
- Легкая форма. Взрослый человек умирает, не дожив до старости.
Несмотря на общий диагноз, β-талассемия у людей может значительно различаться результатами исследований, проявлениями признаков, клиническим течением, лечением и прогнозами на будущее.
Симптомы и проявления
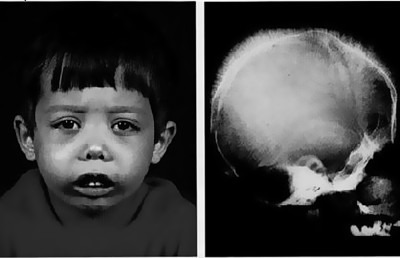
Признаки зависят от формы и времени появления мутации. Большая талассемия случается при гомозиготном происхождении, проявляется у детей сразу после рождения:
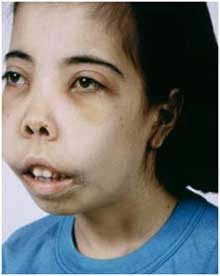
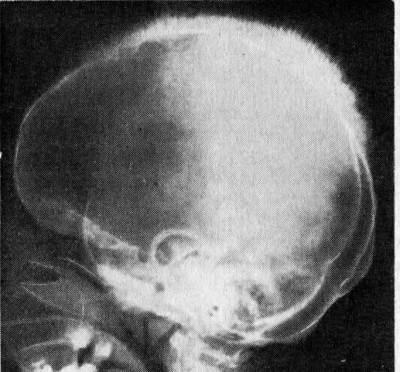
- Удлиненная кверху черепная коробка.
- Верхняя челюсть больше развита по сравнению с нижней.
- Лицевой скелет черепа напоминает монголоидный.
К году жизни наблюдаются следующие признаки:
- Расширение перегородки носа, приплюснутая его форма.
- Формирование аномальных костных наростов в области стоп.
- Нарушения прикуса.
- Желтушность кожных покровов из-за нарушения функции селезенки.
Дефицит кислорода в тканях вызывает такие симптомы:
- Увеличение печени, развитие раннего цирроза.
- Формирование сердечной недостаточности. Излишек железа скапливается на мышце миокарда и нарушает сократительную деятельность мышцы сердца.
- Запаздывание в физиологическом и интеллектуальном развитии.
- Формирование сахарного диабета.
У взрослых людей проявляются следующие симптомы:
- Формирование язв на кожных покровах, их провоцируют аномальные нарушения в кровообращении.
- Частые пневмонии.
- Регулярные переломы костей.
- Воспалительные процессы в желчном пузыре, связанные с отложением камней.
- Кардиосклероз — патология сердца, характеризующаяся разрастанием соединительного рубца в миокарде, замещением мышц и деформацией клапанов.
- Формирование сепсиса при инфекционных патологиях.
- Нарушения на половом уровне.
При развитии промежуточной формы заболевания не наблюдается изменений внешнего вида человека, умственное и физическое развитие в норме, однако есть увеличение селезенки и ломкость костей.
Признаки большой талассемии
Наблюдается сразу после появления на свет:
- Удлиненная форма черепа.
- Монголоидный тип лица.
- Увеличение верхней челюсти.
- Увеличение перегородки носа.
Проведение анализа крови указывает на гепатомегалию, которая заканчивается формированием цирроза и диабета.
.jpg)
Если при рождении ставится диагноз “талассемия”, причем признаки яркие, то чаще всего такие дети могут прожить около года.
Симптомы малой талассемии
Если генная мутация передалась от одного из родителей, то развивается малая талассемия. Симптомы в таком случае размытые либо не проявляются вообще. Клиническая картина следующая:
- Повышенная утомляемость.
- Ухудшение работоспособности.
- Регулярные боли в голове, беспричинные головокружения.
- Бледность кожи с проявлением желтушности.
- Увеличение селезенки.
Наибольшая опасность такого состояния заключается в повышении риска попадания инфекции в организм.
Диагностика заболевания
Обследование включает в себя несколько этапов:
- Сбор анамнеза. Определяются опасные патологии, которые диагностировались у женщины при беременности и могли стать причиной развития патологии.
- Оценка протекания периода вынашивания ребенка.
- Тщательный осмотр врачом: оценивается общее состояние пациента, состояние и цвет кожи, проводится пальпация брюшной области для определения гепатоспленомегалии.
- Опрос родителей либо самого пациента для выяснения выраженности симптомов и тяжести заболевания.
Также назначается проведение следующих лабораторных исследований:
- Биохимический анализ крови.
- ПЦР-тесты.
- Исследование биологического синтеза цепочек гемоглобина.
- Общее исследование крови.
- Электрофорез гемоглобина.
Также необходимо проведение инструментальных методов диагностики:
- УЗИ брюшной полости (обнаруживает увеличение селезенки).
- Рентген костей.
- Пункция костного мозга.
- Развитие плода в утробе матери.
При обнаружении гомозиготной формы патологии назначается искусственное прерывание беременности.
Лечение
Определив, что это такое и каковы признаки, необходимо выбрать методы терапии. Лечебные меры в первую очередь направляют на нормализацию количества гемоглобина.
Помимо этого, есть и другие методы лечения:
.jpg)
- При тяжелых формах недуга необходимо проводить частое переливание крови, но это дает временный результат.
- Назначается хелат железа.
- В современной медицине есть возможность проводить переливание размороженных либо отфильтрованных эритроцитов. Такая процедура более безопасна для детей.
- При увеличенной селезенке назначается ее удаление. Хирургическое лечение возможно только после 5 лет жизни, необходимо учитывать возрастание риска проникновения инфекции.
- Трансплантация костного мозга, но найти донора проблематично.
К клиническим рекомендациям относится специальная диета, которая подразумевает прием орехов, чая, сои, какао (продуктов, которые противостоят усвоению железа). Прием аскорбиновой кислоты, которая способствует выводу железа из организма.
Возможные осложнения
При умеренных и тяжелых формах лечебные меры направлены на продление периода жизни и устранение возможных осложнений, таких как:
- Патологии сердца и печени. Высокий уровень железа в организме может стать причиной повреждения различных тканей, больше всего этому подвержены сердечная мышца и печень. Это нередко становится причиной смертельного исхода, поэтому необходимо проведение регулярного переливания крови.
- Проникновение инфекции в организм также нередко является причиной смерти пациента, особенно это касается тех, у кого проводилась операция по удалению селезенки.
- Остеопороз. При такой патологии костные элементы подвержены частым переломам.
Профилактика
Вылечить талассемию невозможно, поэтому необходимо принимать меры профилактики, которые заключаются в следующем:
.jpg)
- Осуществление диагностики во время беременности, особенно если у обоих родителей наблюдается талассемия.
- Проведение генетического исследования.
- При необходимо проводится искусственное прерывание беременности.
Прогнозы
При диагностировании малой талассемии прогнозы хорошие, человек ведет нормальную жизнь, продолжительность которой практически как у здорового.
При бета-талассемии лишь некоторые больные способны дожить до половозрелого возраста.
Тяжелая гомозиготная форма требует постоянного лечения, поскольку только такие мероприятия, как переливание крови, поддерживают жизнь пациента.
| Талассемия | |
|---|---|
| МКБ-10 | D 56 56. |
| МКБ-10-КМ | D56 и D56.9 |
| МКБ-9 | 282.4 282.4 |
| МКБ-9-КМ | 282.4 [1] [2] , 282.40 [1] [2] и 282.49 [2] |
| MedlinePlus | 000587 |
| eMedicine | ped/2229 |
| MeSH | D013789 и D013789 |
Талассеми́я (анемия Кули) — заболевание, наследуемое по рецессивному типу (двухаллельная система), в основе которого лежит снижение синтеза полипептидных цепей, входящих в структуру нормального гемоглобина. В норме основным вариантом (97 %) гемоглобина взрослого человека является гемоглобин А. Это тетрамер, состоящий из двух мономеров α-цепей и двух мономеров β-цепей. 3 % гемоглобина взрослых представлено гемоглобином А2, состоящим из двух альфа- и двух дельта-цепей. Существуют два гена HBA1 и HBA2, кодирующих мономер альфа, и один HBB-ген, кодирующий мономер бета. Наличие мутации в генах гемоглобина может привести к нарушению синтеза цепей определённого вида.
Содержание
Классификация [ править | править код ]
В зависимости от того, синтез какого из мономеров нарушен, разделяют альфа-, бета- и дельта-талассемию. По тяжести клинических проявлений выделяют тяжёлую, среднюю и лёгкую формы заболевания.
Альфа-талассемия [ править | править код ]
Связана с мутациями в генах HBA1 и HBA2. Есть всего 4 локуса, кодирующего α-цепи. Наличие мутации в одном из локусов приводит к минимальным клиническим проявлениям. Нарушения в двух локусах выражаются лёгкой формой анемии. При мутациях в трёх локусах возникает значительное уменьшение продукции α-глобина. При этом избыточные цепи β-глобина образуют тетрамеры — гемоглобин Н. Эта форма носит также название гемоглобинопатии Н. Характер заболевания может варьироваться от лёгкой до тяжёлой картины гипохромной микроцитарной анемии. Присутствие мутаций во всех четырёх аллелях альфа-глобина не совместимо с жизнью. Ребёнок с такой патологией погибает внутриутробно или вскоре после рождения. Из пуповинной крови таких детей можно выделить гемоглобин Барта.
Бета-талассемия [ править | править код ]
Существует два варианта бета-талассемии — большая талассемия CD8(-AA) и малая талассемия (minor), из которых большая талассемия — наиболее тяжёлая форма заболевания. Возникает при наличии мутаций в обоих аллелях гена бета-глобина. В отсутствие или при резком уменьшении производства бета-цепей гемоглобин А вытесняется гемоглобином F, в норме вырабатывающимся у плода и сменяющимся на гемоглобин А после родов. Малая талассемия связана с наличием мутации в одном из аллелей гена бета-глобина. Как правило, протекает легко и не требует лечения.
Этиология [ править | править код ]
Талассемию вызывают точечные мутации или делеции в генах гемоглобина, ведущие к нарушению синтеза РНК, что приводит к уменьшению или полному прекращению синтеза одного из видов полипептидных цепей. Синтез цепей другого вида продолжается. Это приводит к образованию нестабильных полипептидных агрегатов из избыточных цепей, нарушающих нормальное функционирование эритроцитов, и их разрушению. Повышенный гемолиз эритроцитов вызывает анемию.
Эпидемиология [ править | править код ]
Альфа-талассемия распространена в Западной Африке и Южной Азии. Бета-талассемия часто встречается в странах Средиземноморья, Западной Азии и Северной Африки. Это регионы, где распространена малярия. Гетерозиготные носители мутаций в генах альфа- и бета цепей гемоглобина являются более устойчивыми к малярийному плазмодию. Имеются очаги талассемии в Азербайджане, в равнинных районах которого гетерозиготная бета-талассемия наблюдается у 7—10 % населения.
Клиническая картина [ править | править код ]
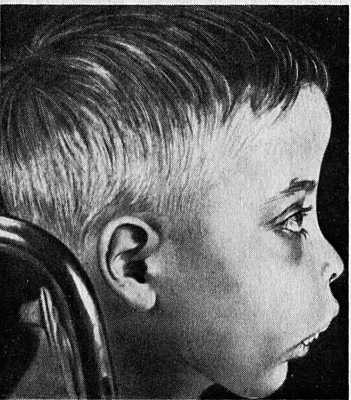
При талассемии характерны гипохромная анемия, анизоцитоз эритроцитов, наличие мишеневидных форм эритроцитов (пятно гемоглобина в центре клетки, напоминающее мишень). При этом содержание сывороточного железа нормальное или повышенное. Компенсаторная гиперплазия костного мозга ведёт к нарушениям в строении лицевого черепа. Череп может стать квадратным, башенным; нос приобретает седловидную форму; нарушается прикус и расположение зубов. Отмечается желтушность кожи и слизистых оболочек. Селезёнка и печень увеличены. Больные подвержены инфекционным заболеваниям. Рано начавшаяся анемия обуславливает физическое и умственное недоразвитие ребёнка.
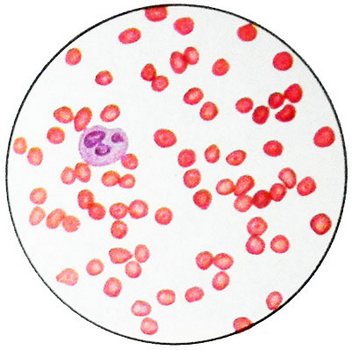
Гемоглобин – элемент эритроцитов, его задача – транспортировать кислород. В самом общем виде кровь состоит из альфа- и бета-белков. Эритроциты не производятся в нужном количестве и не могут перенести достаточный объем кислорода, когда организм не вырабатывает удовлетворительное число одного из этих белков. Результатом является анемия, развивающаяся с рождения и длящаяся на протяжении всей жизни.
Классификация
Различают такие виды, как α- и β-талассемию.
Альфа-талассемия
 В формировании альфа-цепи гемоглобина участвуют 4 гена (по 2 от каждого из родителей).
В формировании альфа-цепи гемоглобина участвуют 4 гена (по 2 от каждого из родителей).
- Если мутация в 1 гене — симптомов болезни нет. Но человек является носителем заболевания, есть вероятность того, что он передаст болезнь ребенку;
- Мутировало 2 гена – лёгкая форма заболевания (малая альфа-талассемия, гетерозиготная талассемия);
- 3 гена – проявление симптомов будет умеренным. Эта форма называется гемоглобинопатия H (hemoglobin H disease);
- Четыре гена – альфа-талассемия майор (alpha-thalassemia major, hydrops fetalis). Состояние приводит к выкидышу, или ребенок погибает почти сразу после родов.
Бета-талассемия
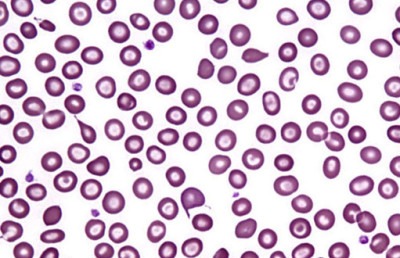 При талассемии этого вида в формировании бета-цепи участвуют 2 гена (один от каждого родителя).
При талассемии этого вида в формировании бета-цепи участвуют 2 гена (один от каждого родителя).
- Мутация одного гена – малая бета-талассемия (beta-thalassemia minor,beta-thalassemia trait). Лёгкая форма заболевания.
- Мутация двух генов свидетельствует о том, что проявления болезни будут сильными. Это бета-талассемия майор (большая талассемия, beta-thalassemia major) или анемия Кули (Cooley’s anemia). Дети такими генами здоровы в первые дни жизни, но в последующие два года у них развиваются симптомы болезни.
Причины заболевания
Талассемия проявляется из-за мутаций в ДНК клеток, которые составляют гемоглобин. Эти мутации нарушают нормальную производительность гемоглобина и влекут за собой снижение его уровня, вызывая анемию. Это генетический дефект, наследуемый ребенком от родителей. Если один из родителей – носитель болезни, у ребенка, скорее всего, разовьется талассемия минор. Видимых симптомов не будет, но ребенок станет носителем заболевания. Некоторые люди с малой талассемией замечают незначительные симптомы.
Про талассемию и риски заболеть в видео ниже
Анна Поняева. Закончила нижегородскую медицинскую академию (2007-2014) и Ординатуру по клинико-лабораторной диагностике (2014-2016).Задать вопрос>>
Симптомы и признаки
 Повышенная усталость;
Повышенная усталость;- Слабость во всём теле (из-за снижения уровня гемоглобина);
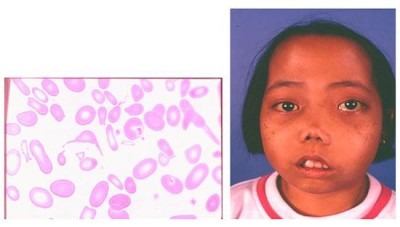
- Бледность кожных покровов, изменение цвета кожи на жёлтый (на фото имеет ярко-жёлтый оттенок);
- Дефекты лицевого скелета;
- Замедленное развитие;
- Скопление газов, вздутие, метеоризм;
- Темно-коричневая моча.
Эти симптомы напрямую зависят от вида и тяжести болезни. Клиническая картина талассемии колеблется в широких диапазонах в зависимости от тяжести состояния и возраста пациента на момент постановки диагноза.
 Признаки и симптомы разных типов болезни можно дополнить следующими:
Признаки и симптомы разных типов болезни можно дополнить следующими:
- Альфа-талассемия майор: бледность, вздутый живот, явные гематологические нарушения у новорожденных;
- Бета талассемия минор: состояние гемолитической анемии, сильная бледность, вздутый живот из-за гепатоспленомегалии;
- Резкие костные изменения из-за неэффективного производства эритроцитов (гиперостоз лобной кости, видимые лицевые кости, изменение зубного прикуса);
- Гиперметаболизм;
- Подагра в связи с гиперурикемией (редко);
- Слишком большое количество железа;
- Замедление роста и развития;
- Метаболические симптомы (сахарный диабет, заболевания щитовидной железы);
- Невропатия / паралич у больных с тяжелой анемией.
Диагностика
Проводится путём проведения исследований, таких как диагностика костного мозга, электрофорез гемоглобина, анализ крови на гемоглобин. Общего анализа крови, как правило, достаточно, чтобы начать подозревать болезнь.
Задача генетических исследований семьи – показать, является ли пациент носителем.
Пренатальная диагностика проводится на 11 неделе беременности путем взятия пробы части плаценты. Плод может быть протестирован при помощи анализа околоплодных вод (амниоцентеза) на 16-й недели беременности. Во время процедуры используется специальная игла, чтобы взять пробу жидкости, окружающей ребенка, для тестирования.
Существует новая методика предимплантационной генетической диагностики (ПГД), она используется в сочетании с экстракорпоральным оплодотворением, её основная задача – помощь родителям-носителям в рождении здоровых детей.
Разновидности альфа-талассемии
 Silentcarrierstateили альфа-талассемия минор (малая альфа-талассемия). Состояние не влияет на здоровье. Недостаток альфа-белка невелик настолько, что не вызывает нарушений функций гемоглобина. Талассемия называется малой (silentcarrier– тихий носитель) из-за затруднительной постановки диагноза. Он ставится родителю, если у ребенка есть проявления гемоглобинопатии H.
Silentcarrierstateили альфа-талассемия минор (малая альфа-талассемия). Состояние не влияет на здоровье. Недостаток альфа-белка невелик настолько, что не вызывает нарушений функций гемоглобина. Талассемия называется малой (silentcarrier– тихий носитель) из-за затруднительной постановки диагноза. Он ставится родителю, если у ребенка есть проявления гемоглобинопатии H.
Гемоглобинопатия ConstantSpring (CS). Форма предыдущего состояния, вызванная изменением альфа-глобина. Название ConstantSpring получила в честь региона Ямайки, где впервые была выявлена. Пациент с CS не ощущает симптомов этого типа, осложнений нет.
Альфа-талассемия минор (minor, малая альфа-талассемия). Значительный недостаток альфа-глобина. Маленький размер эритроцитов и слабовыраженные признаки анемии – частые симптомы этого типа.
Гемоглобинопатия H. Значительный дефицит альфа-глобина, вызывающий тяжелую анемию и серьезные осложнения (спленомегалия селезенки, деформация конечностей). Её назвали в честь гемоглобина H, разрушающего эритроциты.
Гемоглобинопатия Н Constant Spring. Тяжёлая форма. У пациентов отмечены серьёзные признаки анемии. Этому состоянию присущи очень длинные цепи гемоглобина.
Гомозиготная талассемия Constant Spring. 2 постоянных носителя (родители) гемоглобинопатии HCS передают гены талассемии ребенку (в отличие от гемоглобинопатии H CS, при которой один родитель – носитель гемоглобинопатии HCS, а другой – переносчик альфа-талассемии минор).
Водянка плода (альфа-талассемия майор, major). Альфа-белки полностью отсутствуют. Гамма-глобины, производимые плодом, образуют аномальный гемоглобин Barts. Большая часть детей с этим заболеванием умирают еще до родов. В крайне редких случаях переливания крови позволяли родить ребёнка талассемией майор, когда заболевание было диагностировано в утробе, но новорожденный с таким диагнозом в дальнейшем требует постоянных переливаний.
Разновидности бета-талассемии
 Талассемии минор (minor, малая талассемия). Малое количество бета-белка не провоцирует никаких осложнений. Человек является носителем, симптомов, как правило, нет, кроме легкой анемии. Специалисты часто расценивают маленький размер эритроцитов как признак железодефицитной анемии, что в корне неверно.
Талассемии минор (minor, малая талассемия). Малое количество бета-белка не провоцирует никаких осложнений. Человек является носителем, симптомов, как правило, нет, кроме легкой анемии. Специалисты часто расценивают маленький размер эритроцитов как признак железодефицитной анемии, что в корне неверно.
Талассемии интермедиа (intermedia). Дефицит бета-белка велик, это способно вызвать симптомы тяжелой анемии, а также серьезные осложнения (спленомегалию селезенки). Существует широкий диапазон клинической тяжести этого состояния, между талассемией интермедиа и талассемией майор довольно размытые границы, что может привести к ошибкам в диагнозе. Решающее значение играет число переливаний крови. Насколько сильно пациент зависит от переливаний, настолько больше будет вероятность, что его состояние граничит с талассемией майор.
Талассемия майор (major, анемия Кули). Самая тяжелая форма. Абсолютное отсутствие бета-белка способствует развитию анемии, несущей угрозу для жизни. Анемия Кули требует постоянных переливаний. Эти регулярные переливания крови (чаще на протяжении всей жизни) приводят к тому, что в организме скапливается избыток железа. Задача врачей – держать эти “перегрузки” под контролем с помощью терапии по удалению избытков железа во избежание отказа органов.
E Бета талассемия. Гемоглобин Е – распространенный аномальный гемоглобин. На фоне Е бета талассемии развивается тяжелая анемия.
Серповидная-бета-талассемия. Комбинация гемоглобина S с бета талассемией. Гемоглобин Sвстречается у людей с серповидно-клеточной анемией (эритроциты изменённой формы — дрепаноциты). Этот вид анемии наследуется как доминантный признак.
Различия типов талассемии в виде таблицы:
α-талассемия:
| Недостающие альфа-гены | Расстройство | Признаки | Другие названия |
| 1 | носитель (silent carrier) | отсутствуют | aльфа-талассемия минима, alpha thalassemia minima |
| 2 | минор | лёгкие | aльфа-талассемия минор, alpha thalassemia minor |
| 3 | гемоглобинопатия H | средние | промежуточная альфа-талассемия, Hemoglobin H disease |
| 4 | майор | тяжёлые | водянка плод, альфа-талассемия майор, alphathalassemiamajor |
β-талассемия:
| Пораженные бета-гены | Расстройство | Признаки | Другие названия |
| 1 | носитель (silent carrier) | лёгкие | b талассемия минор, Beta thalassemia minor |
| 1 | минор | лёгкие | |
| 2 | интермедиа | средние | |
| 2 | майор | тяжёлые | анемия Кули, Cooley’s anemia |
Прогноз течения болезни
Пациенты с умеренной и тяжелой формой имеют хорошие шансы на выживание, их главная задача – придерживаться правильной программы лечения (трансфузионная терапия и профилактика уровней железа с помощью комплексообразующих агентов). Сердечно-сосудистые заболевания из-за избытка железа — основная причина смерти. Трансплантация костного мозга способна излечить больного. Пациентам с талассемией, возможно, потребуется хирургическое вмешательство, чтобы исправить скелетные деформации.
Репортаж про ученных, которые научились бороться с данным заболеванием
Больным требуется постоянная диагностика общего анализа крови и уровней железа. Важно проверять печень (из-за развития гемохроматоза) и сердце, сдавать анализы на вирусные инфекции.
При беременности
Женщины с талассемией имеют высокий уровень бесплодия. Тем не менее, некоторые из них могут забеременеть.
- Если у одного из партнеров есть ген талассемии, есть большая вероятность, что ребенок унаследует болезнь;
- Стресс во время беременности только ухудшит симптомы болезни. Сердце и печень женщины являются наиболее уязвимыми во время беременности, очень важная задача – контроль эндокринной системы;
- Во время беременности объем крови в организме значительно возрастает. Это может привести к анемии, которая увеличит потребность в переливании крови. Чем выше объем крови в организме матери, тем больше нагрузка на её сердце;
- Женщины с талассемией имеют повышенный риск развития сахарного диабета 1 типа;
- Во время беременности женщине потреблять достаточное количество фолиевой кислоты, она поможет снизить риск развития мегалобластной анемии.
Заболевание у детей
 У некоторых детей с легкой формой талассемии симптомы могут отсутствовать. Но обычно больные дети чувствуют себя усталыми, раздраженными, у них частая одышка, головокружения, бледная кожа, бледные (синие) губы.
У некоторых детей с легкой формой талассемии симптомы могут отсутствовать. Но обычно больные дети чувствуют себя усталыми, раздраженными, у них частая одышка, головокружения, бледная кожа, бледные (синие) губы.
В более тяжелых случаях, у детей наблюдаются:
- Частый пульс;
- Увеличенная печень или селезенка;
- Желтуха;
- Деформация лобной кости;
- Замедленный рост (из-за анемии).
В более позднем возрасте у детей отмечены болезни сердца, хрупкие кости (остеопороз).
Дети также могут быть диагностированы до рождения, путем проверки образца амниотической жидкости или клеток из плаценты. Дети сдают анализы в том же порядке, что и взрослые, в том числе, анализ крови, анализ на гемоглобин.
Профилактика
- Переливание крови – введение эритроцитов, чтобы восстановить их уровень. Переливания проводятся с периодичностью в каждые 4 месяца (для больных тяжелой талассемией), и каждые 3-6 недель у больных бета-талассемией. Иногда переливания могут потребоваться больным гемоглобинопатией Hили бета-талассемией интермедиа;
- Удаление избытка железа из организма. Опасность множественных переливаний – рост уровня железа в организме, что может стать причиной болезней сердца. Лекарственное средство, например, жидкий дефероксамин вводят под кожу, или больной принимает таблетки, например, Deferasirox. Таким пациентам необходимо принимать добавки с фолиевой кислотой для восстановления эритроцитов;
- Спленэктомия (удаление селезенки) — вынужденная мера для пациентов с гемоглобинопатией Н, если их потребность в переливании постоянно растет;
- Трансплантация костного мозга (и стволовых клеток) от совместимого донора – единственный метод, способный излечить больного. Пересадка костного мозга от родственника даст лучшие результаты. Уже в течение двух недель пересаженные стволовые клетки начнут производить новые, здоровые клетки крови.
Образ жизни при данном заболевании
- Стоит задача избегать потребления большого количества железа. Не стоит принимать витамины или другие добавки, содержащие железо, не проконсультировавшись с лечащим врачом;
- Ешьте здоровую пищу. Сбалансированный ежедневный рацион поможет почувствовать себя лучше. Лечащий врач может рекомендовать добавки с фолиевой кислотой (способствует созданию новых эритроцитов). Чтобы сохранить здоровье костей, рацион должен содержать оптимальное количество кальция и витамина D;
- Избегайте инфекций (в особенности после спленэктомии). Защитить себя от инфекций можно с помощью частого мытья рук и избегая больших скоплений людей;
- Больным талассемией показано прививаться от гриппа, пневмококка и гепатита B, важна диагностика показателей крови.
Группы риска
Частота возникновения альфа талассемии среди белых людей низкая. Около 15% афроамериканцев являются её носителями, и только 3% имеют слабо выраженные признаки. Гемоглобинопатия H редко встречается в этой группе населения. В Северной Америке из-за присутствия большого количества мультинациональных слоёв общества частота заболевания синдромом альфа талассемии увеличилась. У некоторых этнических групп (население Юго-Восточной Азии и средиземноморского населения) гемоглобинопатия H встречается чаще. Высокая частота проявления гемоглобинопатии Constant Spring (CS) была отмечена в Юго-Восточной Азии.
Наследственность
Это заболевание наследуется как не полностью доминантный аутосомный признак от родителей к детям через мутировавшие гены гемоглобина.
Если в семье присутствовали случаи талассемии, ребенок имеет повышенный риск развития заболевания.
Международная статистика
Альфа-талассемия – наиболее распространённое генетическое заболевание в мире. Существует 270 миллионов носителей мутировавших генов глобина, которые могут передать тяжелые формы талассемии по наследству. Кроме того, 300000-400000 больных младенцев рождаются каждый год, более 95% из которых находятся в Азии, Индии или на Ближнем Востоке.
Частота проявления этой болезни составляет 20-30% в частях Западной Африки, 60-80% — в некоторых районах Саудовской Аравии, Индии, Таиланда, Папуа-Новой Гвинеи и Меланезии. В Таиланде около 7000 детей ежегодно рождается с гемоглобинопатией H. Среди китайского населения диапазон больных составил от 5% до 15%. Частота возникновения альфа-талассемии в Великобритании, Исландии и Японии ниже 0,01%.
Пол
Мужчины и женщины затронуты в равной степени.